Immune evasion and provocation by Mycobacterium tuberculosis
.
Mycobacterium tuberculosis, the causative agent of tuberculosis, has infected humans for millennia. M. tuberculosis is well adapted to establish infection, persist in the face of the host immune response and be transmitted to uninfected individuals. Its ability to complete this infection cycle depends on it both evading and taking advantage of host immune responses. The outcome of M. tuberculosis infection is often a state of equilibrium characterized by immunological control and bacterial persistence. Recent data have highlighted the diverse cell populations that respond to M. tuberculosis infection and the dynamic changes in the cellular and intracellular niches of M. tuberculosis during the course of infection. M. tuberculosis possesses an arsenal of protein and lipid effectors that influence macrophage functions and inflammatory responses; however, our understanding of the role that specific bacterial virulence factors play in the context of diverse cellular reservoirs and distinct infection stages is limited. In this Review, we discuss immune evasion and provocation by M. tuberculosis during its infection cycle and describe how a more detailed molecular understanding is crucial to enable the development of novel host-directed therapies, disease biomarkers and effective vaccines.
Introduction
Mycobacterium tuberculosis is a respiratory pathogen that is estimated to infect one-quarter of the world’s population, and it has killed more people over the course of human history than any other microorganism. M. tuberculosis is thought to have originated from environmental mycobacteria that entered human populations in the Horn of Africa more than 70,000 years ago, and the global lineages of M. tuberculosis today mirror the subsequent routes of human migration1. Having evolved with humans for millennia, M. tuberculosis is exquisitely well adapted to navigate the human immune system. The life cycle of M. tuberculosis (Fig. 1) depends on its ability to interact with the immune system in seemingly distinct ways: it evades the innate immune response, persists in the face of an adaptive immune response without causing symptomatic disease, and elicits a robust inflammatory response to cause extensive tissue pathology for it to be transmitted. Understanding how M. tuberculosis orchestrates its life cycle is crucial for the design of preventive and therapeutic vaccines, as well as novel therapies and disease biomarkers.
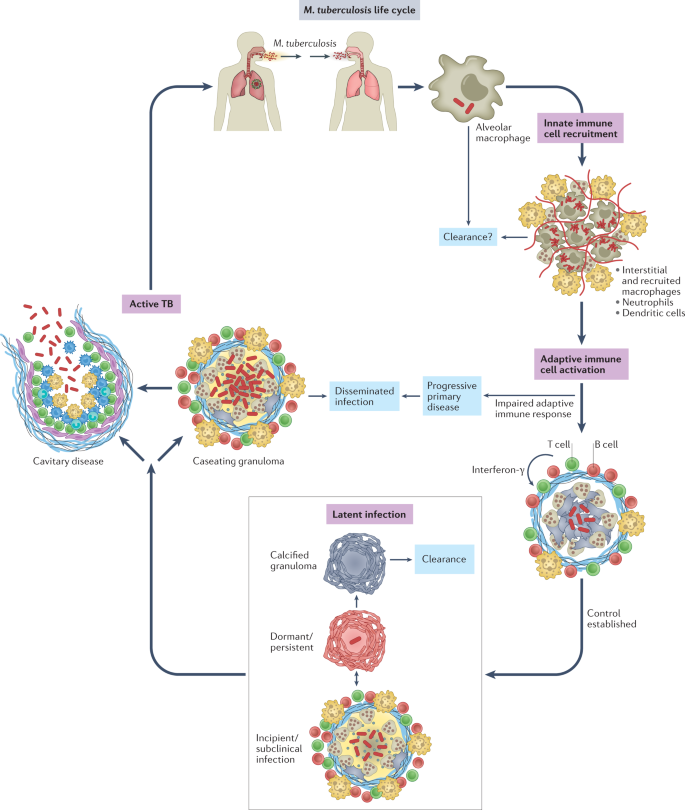
A central feature of tuberculosis (TB) pathogenesis is the ability of the causative organism, M. tuberculosis, to survive in diverse intracellular environments within a variety of myeloid cell populations. The outcome of M. tuberculosis infection is shaped by host genetics, co-morbidities, environmental factors and microbial virulence determinants in ways we are just beginning to understand. To establish infection, M. tuberculosis resists and disarms macrophages and neutrophils in the lung, undermining lysosomal trafficking pathways to survive intracellularly. M. tuberculosis infects tissue-resident alveolar macrophages (AMs), which arise during embryogenesis, as well as a variety of phenotypically distinct macrophage populations of haematological origin2,3,4,5,6,7. Infected dendritic cells (DCs) travel to the draining lymph node and prime T cells, which then return to the infected lung. This takes several weeks, but once an effective adaptive immune response develops, T cells, B cells and activated macrophages form the characteristic granuloma, and bacterial control is established. Most often, bacterial replication is contained, and the inflammatory response subsides, leading to latent TB. Individuals with latent TB have an adaptive immune response to M. tuberculosis but no symptoms, culturable bacilli or disease manifestations. Latent TB likely encompasses a spectrum of outcomes that include bacterial elimination and subclinical disease8, although currently there is no way to distinguish individuals who sterilized infection from those that harbour viable bacilli. Approximately 5–10% of infected individuals will go on to develop active TB, due to either progressive primary infection or ‘reactivation’, which can occur long after initial infection9. In reactivation, TB develops in the setting of a previously ‘successful’ (although not sterilizing) adaptive immune response. A wide variety of disease manifestations are possible, from cavitary lung disease to focal infection involving nearly any organ system, to widely disseminated infection. Cavitary lung disease is most common, and, importantly, individuals with cavitary lesions are the most infectious. Thus, although disseminated infection can be devastating for the infected individual, it is not a successful outcome for the bacteria either, as it is much less likely to lead to transmission. In this Review, we discuss how M. tuberculosis evades immune-mediated clearance while capitalizing on the host inflammatory response at different phases of its life cycle. We focus on recent studies, highlight gaps in knowledge and consider how our current understanding will inform new therapies, vaccines and diagnostics.
How M. tuberculosis establishes infection
The cellular niche of M. tuberculosis
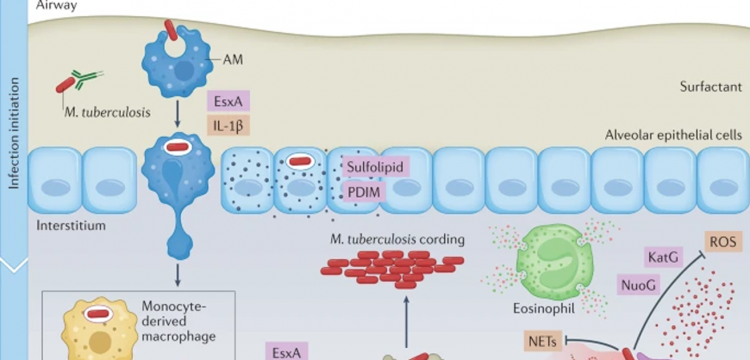
Understanding the earliest events of infection is crucial for the development of a preventive vaccine. The infectious dose of M. tuberculosis is remarkably low, estimated to be approximately three bacilli, highlighting how effective M. tuberculosis is at evading the innate immune response10. Even before uptake by phagocytic cells, M. tuberculosis encounters alveolar lining fluid (Fig. 2), a complex mixture of lipids and proteins secreted by alveolar epithelial cells, which includes surfactant proteins and hydrolases that interact with mycobacterial surface glycolipids. Alveolar lining fluid enhances pathogen uptake and killing by phagocytes and has a variable impact on interactions with alveolar epithelial cells11,12. Deficiencies in pulmonary surfactant due to inflammageing or smoking promote intracellular M. tuberculosis replication and increase the risk of developing TB12,13. In addition, antibody opsonization may promote innate immune control against M. tuberculosis14. Interestingly, recent data suggest that M. tuberculosis-specific IgM antibodies elicited by vaccination might be protective15. This indicates that it might be possible to develop vaccines that generate humoral responses that disrupt the earliest steps of infection, for example, by neutralizing secreted or cell envelope-associated virulence factors or by functionally altering subsequent macrophage interactions, as discussed further herein.
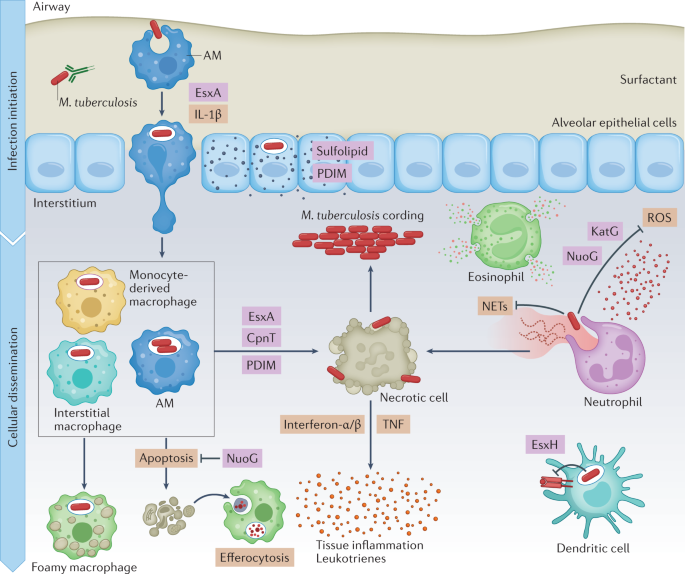
Much of our knowledge of the earliest events of infection come from the mouse model. In that model, epithelial cells are infected in the first 48 h after infection; however, M. tuberculosis does not appear to replicate or persist in these cells7. The mycobacterial lipid phthiocerol dimycocerosate (PDIM) can spread into epithelial membranes and modulate immune responses16. Microfold cells (M cells) may also be a portal of entry, allowing M. tuberculosis to gain access to underlying lymphoid tissue in the upper airways17,18. In mice, tissue-resident AMs are the predominant cell type infected during the first 2 weeks7. AMs are located in the air spaces, where they are continually exposed to environmental particulates. In the absence of a microbial challenge, AMs are poised to suppress inflammatory responses to foreign material to prevent lung injury19,20. M. tuberculosis infection of AMs initiates a nuclear factor erythroid 2-related factor 2 (NRF2)-driven antioxidant transcriptional response that correlates with impaired control of M. tuberculosis growth21,22. Ageing may also make AMs more permissive to M. tuberculosis replication23. After 2 weeks, infected AMs migrate out of the alveolar space into the lung interstitium in a process dependent on host IL-1β signalling and an M. tuberculosis type VII secretion system, ESX-1 (ESAT-6 secretion system 1; ESAT-6 is also known as EsxA), which is discussed later. After entering the lung interstitium, M. tuberculosis infects additional phagocytic cell types7. On the basis of its transcriptional responses, M. tuberculosis within AMs appears to be able to access host iron and fatty acids, experiences minimal oxidative and nitrosative stress, and has a high replicative capacity2,4. Moreover, selective depletion of AMs decreases lung M. tuberculosis burden in mice2,24, supporting the idea that AMs are a particularly permissive niche that facilitates the establishment of infection. However, by 3 weeks after infection, infected AMs can exhibit a pro-inflammatory response5. Recent single-cell RNA sequencing analyses revealed that there are multiple AM subpopulations after infection, some of which mount a pro-inflammatory response and impose stress on the bacilli3. In addition, another study implicated AMs as an M. tuberculosis-restrictive cell type following their migration out of the airways into granulomas25. Overall, the data support the idea that AMs exert little control over M. tuberculosis during the first 2 weeks of infection; however, some AM populations may be restrictive, particularly as the infection progresses and an adaptive immune response develops.